商机详情 -
西藏地拉罗司
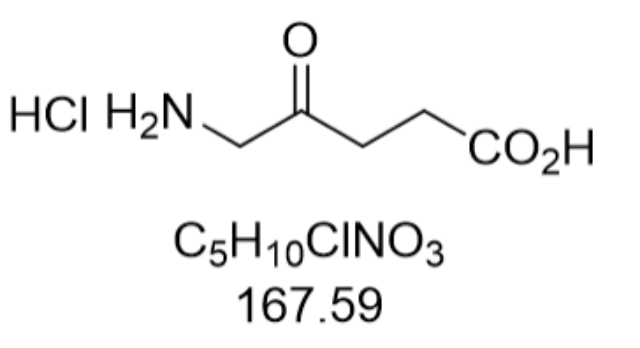
5-氨基乙酰丙酸盐酸盐(5-Aminolevulinic Acid HCl,CAS:5451-09-2)是一种具有独特化学结构的有机化合物,其分子式为C₅H₁₀NO₃Cl,分子量167.59。该物质以白色或淡黄色结晶形态存在,易溶于水,在酸性和中性条件下稳定,但对热和光敏感,需在2-8℃低温避光环境中密封保存。其化学结构中的氨基(-NH₂)和羧基(-COOH)赋予其两性离子特性,而盐酸盐的存在则增强了其水溶性和稳定性。作为生物体内天然存在的非蛋白质氨基酸,5-ALA是血红素、叶绿素及维生素B₁₂等四吡咯类化合物生物合成的关键前体,这一特性使其在生命科学领域具有不可替代的地位。其合成工艺涉及丙二酸酯缩合、亚硝化还原及水解等步骤,通过优化反应条件可实现总收率提升,为工业化生产提供了经济可行的方案。原料药清洁生产工艺是发展趋势。西藏地拉罗司
德兰佐米(Delanzomib,CAS号:847499-27-8)作为第二代口服蛋白酶体抑制剂,其重要性能源于对蛋白酶体糜蛋白酶样活性的精确抑制。该化合物通过靶向20S蛋白酶体的β5亚基,以3.8 nM的IC50值高效阻断其催化功能,较第1代抑制剂硼替佐米(Bortezomib)的抑制强度提升近2倍。实验数据显示,在多发性骨髓瘤(MM)细胞系RPMI-8226中,德兰佐米处理12-24小时即可诱导裂解的caspase-3、-7、-9蛋白表达,48小时内引发细胞凋亡率达65%,而硼替佐米在同等条件下需72小时才能达到50%的凋亡率。这种快速起效特性与其独特的硼酸酯结构密切相关——通过共价结合蛋白酶体活性位点的苏氨酸残基,形成稳定的抑制剂-酶复合物,明显延长靶标抑制持续时间。在SCID小鼠移植瘤模型中,7.8 mg/kg剂量口服给药后,疾病组织蛋白酶体活性抑制率持续维持85%以上达72小时,而正常肝组织抑制率只30%,这种疾病选择性的药效学特征为临床安全用药提供了重要依据。吉林紫杉醇原料药的仿制药生产需通过一致性评价,确保疗效一致。
苯丁酸氮芥属于细胞毒类的化疗药物,主要通过影响疾病细胞的DNA功能来实现抗疾病医治的目的。作为一种口服制剂,它主要用于医治慢性淋巴细胞性白血病、低度恶性非霍奇金淋巴瘤和卵巢疾病等。苯丁酸氮芥的使用也伴随着一定的不良反应,常见的有消化道反应和骨髓抑制。消化道反应包括恶心、呕吐、食欲不振等,而骨髓抑制则可能导致白细胞、血小板和红细胞的减少。因此,在使用苯丁酸氮芥期间,需要严密监测患者的血常规及肝肾功能,一旦出现异常,应及时处理。对于严重骨髓抑制或严重肝肾功能损害的患者,应禁止使用苯丁酸氮芥。由于该药物具有致突变性,可能导致男性染色单体和染色体损害,并可增加急性白血病的发生率,因此在使用时应权衡潜在的医治益处与风险。
在临床医治性能方面,美法仑展现出多靶点抗疾病谱系。对多发性骨髓瘤的疗效确立其医治地位,大剂量化疗(200 mg/m²)联合自体干细胞移植可使5年生存率提升至65%。在实体瘤医治中,动脉灌注技术突破血脑屏障限制,对恶性黑色素瘤肢端型患者局部控制率达82%,软组织肉瘤保肢率提高至75%。特殊剂型开发方面,脂质体包裹技术将较大耐受剂量从常规16 mg/kg提升至40 mg/kg,同时降低骨髓抑制发生率。联合用药研究显示,与硼替佐米联用可使敏感患者中位无进展生存期延长至24个月,与达雷妥尤单抗三药的方案更将完全缓解率推高至62.5%。耐药机制研究指出,谷胱甘肽S-转移酶过表达和DNA修复酶活性增强是主要耐药途径,针对这些靶点的联合用药策略正在临床试验中验证。原料药生产中使用的有机溶剂需合规回收,降低安全隐患。
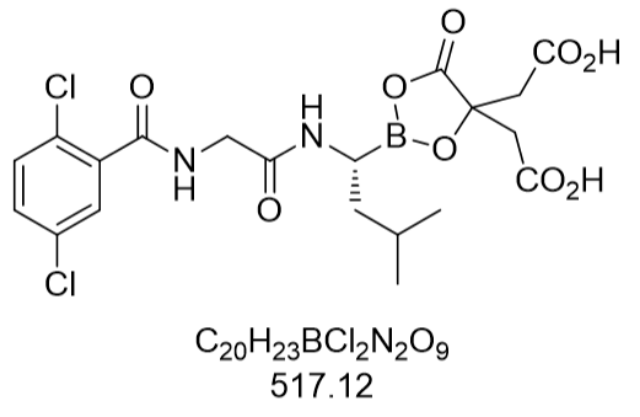
在临床应用层面,艾沙佐咪与来那度胺、的联合方案已成为复发/难治性多发性骨髓瘤的标准医治选择。TOURMALINE-MM1全球Ⅲ期临床试验纳入722例患者,结果显示联合方案组的中位无进展生存期(PFS)达20.6个月,较对照组的14.7个月明显延长。中国亚组分析进一步证实,亚洲患者群体中联合方案组的总缓解率(ORR)达62%,其中完全缓解率(CR)为19%。值得注意的是,该方案采用全口服给药模式,患者只需每周服用1次艾沙佐咪(4 mg),配合每日来那度胺(25 mg)和每周(40 mg),极大提升了医治依从性。2017年《中国多发性骨髓瘤诊治指南》将其列为二线医治选择的方案,2023年更新的NCCN指南更将其纳入维持医治推荐。原料药研发周期长、投入成本高。吉林紫杉醇
原料药的生产工艺研究需结合国际合作,引进先进技术。西藏地拉罗司
从药代动力学特征分析,德兰佐米的口服生物利用度达62%,明显优于硼替佐米的35%,这得益于其优化的分子极性(LogP=2.1)和肠道吸收特性。在C57BL/6小鼠模型中,单次口服10 mg/kg后,血浆Cmax达1.2 μM,T1/2为6.8小时,较静脉注射硼替佐米的3.2小时延长一倍。组织分布研究显示,德兰佐米在疾病组织的AUC0-24h是正常肌肉组织的3.7倍,这种靶向富集效应与其通过EPR(增强渗透与滞留)效应穿透疾病血管内皮密切相关。代谢稳定性方面,该化合物在肝微粒体中的半衰期为45分钟,主要代谢产物M1(去甲基化衍生物)保留85%的母体活性,形成有效的代谢启动循环。值得注意的是,德兰佐米对细胞色素P450酶系的抑制作用较弱(IC50>50 μM),明显降低药物-药物相互作用风险,为联合化疗方案提供安全保障。西藏地拉罗司